PolyFARM Documentation
- The general idea
- The knowledgebase files (.kb)
- The background files (.bg)
- The settings files (.s)
- How to use Farmer, Worker and Merger
- How to use CreateAttributes, RemoveDuplicates
The general idea
The idea is to collect frequent first order patterns from data represented as prolog (actually currently restricted to ground Datalog). A pattern might look like this:
hom(A,B), mol_wt(B,5), classification(B,"gammabacteria").
This pattern has 3 atoms. We say this pattern has been found at level 3. The algorithm iteratively adds more atoms to the patterns, level by level. All level 1 frequent patterns will be produced first, then all level 2 patterns and so on. PolyFARM is desigend to be run as jobs which can be batched and sent to a Beowulf cluster. There are 3 components to PolyFARM:
- Farmer
- Worker
- Merger
The knowledgebase files (.kb)
The knowledgebase files are the files of data you want patterns to be found in. Partition your database into appropriately sized pieces, named filestem1.kb, filestem2.kb, filestem3.kb and so on. filestem is whatever name you want. Each example (transaction/unit of data) in your kb files is separated by an '@' character on a line by itself. For example:
orf(yeast1).
similar(yeast1,p1)
weight(p1,heavy).
similar(yeast1,p2)
weight(p2,heavy).
@
orf(yeast2).
similar(yeast2,p3)
weight(p3,light).
similar(yeast2,p4)
weight(p4,medium).
An '@' is optionally allowed at the end of the file too. Experiment with the size of your kb files until they are small enough to be processed comfortably. See BGKBParser.ly for more detail on the grammar of a kb file.
The background file (.bg)
This contains predicates that are used to generate constants, eg
kword(membrane).and also value hierarchies.
kword(transmembrane).
kword(plasmid).
hierarchy(species,
[bacteria [salmonella,listeria,escherichia],
viruses [simplexvirus,herpesvirus]
]).
Constant-generating predicates come first, then an '@' character by itself on a line, then the hierarchies. Either are optional. Whitespace is not important. Nested lists indicate the parent-child relationships in the hierarchy. See BGKBParser.ly for more detail on the grammar of a bg file.
The settings file (.s)
This contains all user-defined settings.
Many machine learning algorithms allow the user to specify a language bias. This is simply the set of factors which influence hypothesis selection (Utgoff1986). Language bias is used to restrict and direct the search. Weber (Weber1998) and Dehaspe (DehaspePHD) describe more information about declarative language biases for datamining in ILP. Weber describes a language bias for a first order algorithm based on APRIORI. Dehaspe describes and compares two different language biases for data mining: DLAB and WRMODE. We will follow the lead of WARMR and allow the user a WRMODE-like declarative language bias which permits the specification of modes, types and constraints. SettingsParser.ly contains the grammar of a settings file.
Support and confidence
These are expressed as rationals (fractions).
support(2/3).
confidence(1/1).
Modes
Modes express how arguments of literals are to be created. Modes are often used in ILP (and are notorious for being misunderstood by the users!). Arguments of an atom can be specified as + (must be bound to a previously introduced variable), - (introduces a new variable), or as introducing a constant. In PolyFARM, constants can be specified in two ways, which are simply syntactic variants for ease of comprehension:
constlist [ ... ]
, where the list of available constants is given immediately in the mode declaration (and is a short list), and
constpred predname
, where the constants are to be generated by an arity 1 predicate in the background knowledge file (this case would be chosen when the list of constants is large).
For example, the following declares that the sq_len predicate must take an already-existing variable for its first argument, then an integer between 2 and 6 for its second argument:
mode(sq_len(+,constlist [2,3,4,5,6])).
The user can also specify that an argument is a constant taken from a hierarchy (such as a species taxonomy). This is done by the use of the word "consttree", and an appropriate definition of a hierarchy in the background knowlegde. For example, the second argument of "classification" is a constant from the hierarchy specified by "species" in the background knowledge:
mode(classification(+,consttree species)).
Types
Types are used to restrict matching between arguments of literals. An argument which has a mode of +, must be bound to a previous variable, but now we can also ensure that this previous variable must be of the same type.
For example the following declares that the argument of "student" must be the same type as the first argument of "buys":
buys(Person,Food).
student(Person).
Key
The key predicate is not flexible as in WARMR. Rather, it is the predicate that must appear in all associations, the root of the association tree.
Constraints
Constraints are used when further restrictions are required. Currently constraints can be used to restrict the number of times a predicate can be used in any one query, or to state that one predicate may not be added if another predicate is already in the query. The latter form of constraint can be used to ensure that duplicate queries are not produced through different orders of construction. For example, the queries:
buys(pizza,X) and student(X)
and
student(X) and buys(pizza,X)
are equivalent.
Add constraint(x,y) if you don't want x adding to a clause that already has y.
constraint(similar,kword).
constraint(kword,weight).
Add constraint(x,N) if you want to allow x to be used up to N times within an association (the default is once only).
constraint(similar,2).
Rule Head
If association rules (query extensions) are to be generated, then this is used to state which predicate is to be used as the rule head. For example, the following states that the "function" predicate is to be used in the rule head:
rulehead(function).
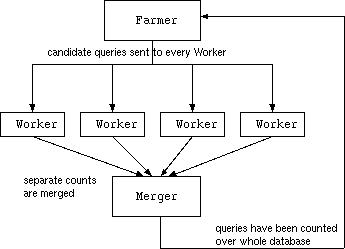
How to use Farmer,Worker and Merger
First of all, run Farmer. Farmer requires 4 arguments: filename level chunks workingdir
- filename: filestem of files to be processed (if you have yeast.s, yeast.bg and yeast1.kb then this is "yeast")
- level: the number of the level to be created (this is 1 initially, then you should increment it each time you run Farmer again). You have a choice here, in case it is necessary to rerun a level due to eg hardware failure, accidental file deletion etc.
- chunks: the maximum number of database chunks that you have. For example, if you have yeast1.kb, yeast2.kb, yeast3.kb and yeast4.kb then this is 4. You may wish to just use 1 or 2 for initial testing.
- workingdir: where the beowulf job file is to be written, candidate associations are to be written and the previous level's merged counts are to be read from.
Farmer generates a list of worker jobs to be run on the Beowulf nodes, and initial candidate queries for the workers to count.
Our Beowulf cluster has a simple scheduler which allows many users to submit jobs that will be processed as fairly as possible. The scheduler was written in TCL and so the list that Farmer produces is currently in the format required by our scheduler software. However it is simply a list of commands that could each be run directly (eg with rsh). For example, this is a list of 4 jobs for the Workers, at level 3:
"/home/ajc99/parallel/Worker /common/ajc99/finalyeast/yeast 3 1 /home/ajc99/parallel/finalyeast/ +RTS -K5M"
"/home/ajc99/parallel/Worker /common/ajc99/finalyeast/yeast 3 2 /home/ajc99/parallel/finalyeast/ +RTS -K5M"
"/home/ajc99/parallel/Worker /common/ajc99/finalyeast/yeast 3 3 /home/ajc99/parallel/finalyeast/ +RTS -K5M"
"/home/ajc99/parallel/Worker /common/ajc99/finalyeast/yeast 3 4 /home/ajc99/parallel/finalyeast/ +RTS -K5M"
Farmer also generates a file of candidate associations for the Workers to count. This will be called Qname.level.data, where name is the base filename, and level is the level number supplied as an argument to Farmer. Farmer also generates a binary file containing the settings and background knowledge needed by the Workers (name.level.data)
Next each of the Worker jobs in the list must be run. Each of these will create a file named Qname.databasePartitionNumber. These files contain the counts for the queries/associations in this partition.
At any time, Merger can be run to collect the results of completed Worker jobs. Merger takes 4 arguments:
- filename
- chunk_from
- chunk_to
- workingdir
This will merge all files of Worker results from chunk_from to chunk_to, inclusive. For example
Merger yeast 11 20 yeastresults
Merger stores these merged results in a file named Qname.merged. WARNING: this file is modified each time Merger is run, and counts are accumulated. After Merger has been run on a set of Worker results then the Worker results files can be deleted to save filespace.
Finally, when all counts have been collected and merged into the Qname.merged file, then the next level of Farmer can be run again. This time Farmer will output the frequent associations found so far, and make a new set of candidates for the Workers to count.
A typical PolyFARM run might look like this:
Farmer yeast 1 3 outputdir
Worker yeast 1 1 outputdir #
Worker yeast 1 2 outputdir # done in parallel on Beowulf nodes
Worker yeast 1 3 outputdir #
Merger yeast 1 3 outputdir
Farmer yeast 2 3 outputdir ## level 1 associations output here
Worker yeast 2 1 outputdir #
Worker yeast 2 2 outputdir # done in parallel on Beowulf nodes
Worker yeast 2 3 outputdir #
Merger yeast 2 3 outputdir
Farmer yeast 3 3 outputdir ## level 2 associations output here
Worker yeast 3 1 outputdir #
Worker yeast 3 2 outputdir # done in parallel on Beowulf nodes
Worker yeast 3 3 outputdir #
Merger yeast 3 3 outputdir
Farmer yeast 4 3 outputdir ## level 3 associations output here
How to use CreateAttributes and RemoveDuplicates
CreateAttributes takes 4 arguments: filename patternfile chunkfrom chunkto
This takes a file of associations (as produced by Farmer) patternfile and chunks of a database and produces lists of boolean values for each example in the database, representing whether each association is present or absent in that example.
RemoveDuplicates takes 2 arguments: filename patternfile
This takes a file of associations patternfile and removes associations that are duplicates or equivalent to each other from this file. It writes the results to stdout. For example an association that appears twice with differently named variables is detected. The test for equivalence of two associations $Q_1$ and $Q_2$ is if both $Q_1$ subsumes $Q_2$ and $Q_2$ subsumes $Q_1$ then they are equivalent, and one of them is unnecessary. At present this stage must be carried out after PolyFARM has finished execution, but it should be added into the main body of the program in future.